
Les syndromes d’Ehlers-Danlos (SED)
Article
Publié le 23/01/2023
Information proposée par UNSED, Union Nationale des Syndromes d’Ehlers-Danlos
Syndromes d’Ehlers-Danlos :
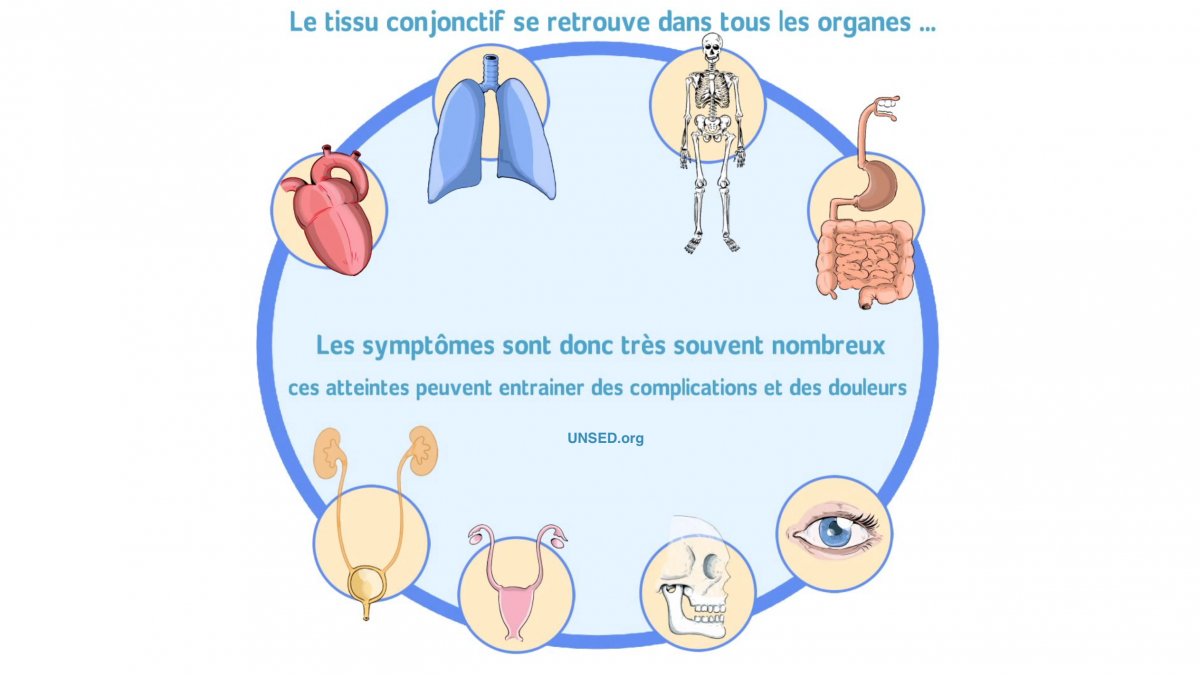
Les syndromes d’Ehlers-Danlos (SED) sont des maladies héréditaires du tissu conjonctif caractérisées par la triade : hyperlaxité articulaire, hyperélasticité cutanée (légère, modérée ou importante selon le type de SED) et fragilité des tissus conjonctifs.
Ils sont essentiellement dus à des anomalies de biosynthèse et/ou de structure de protéines de la matrice extracellulaire.
Leur prévalence d’ensemble en population générale est estimée à 1 pour 5 000, ce qui en fait des maladies rares au sens de la définition européenne d’une maladie rare (prévalence en population générale < 1/2.000).
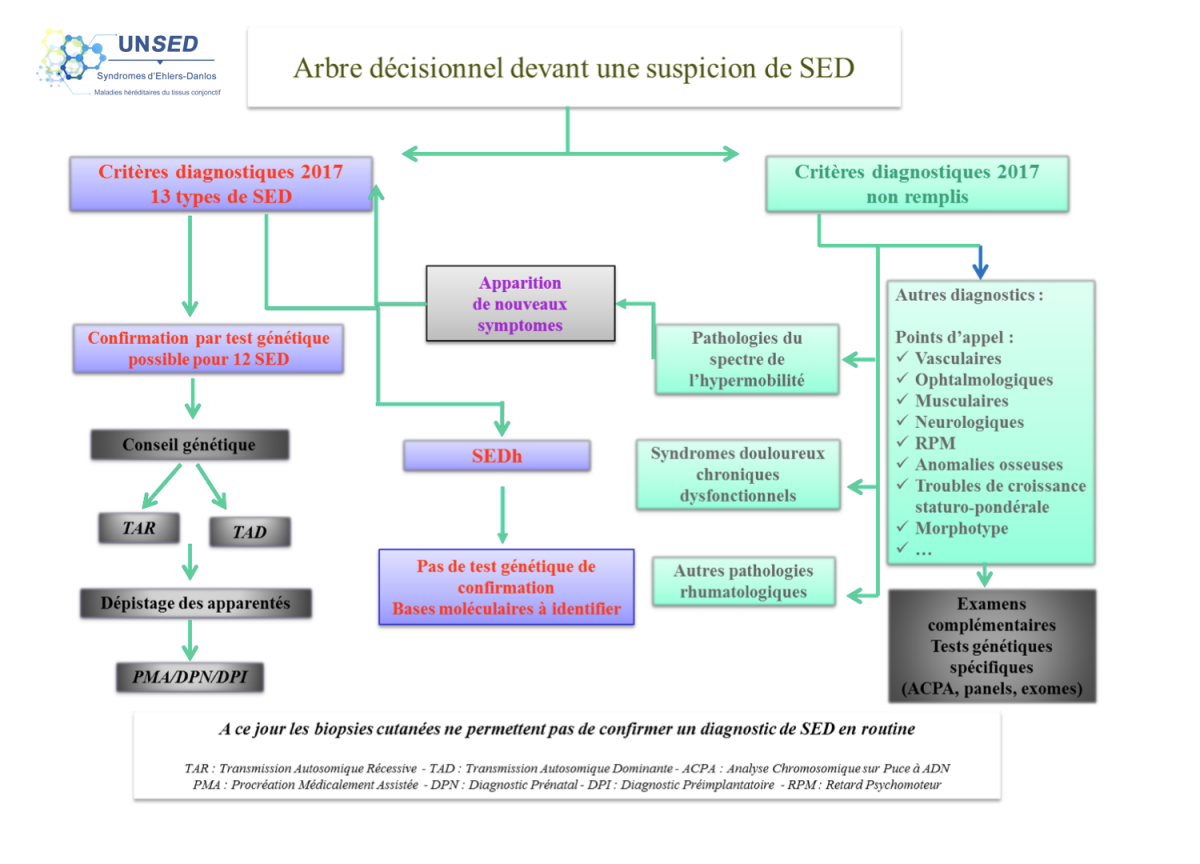
La dernière classification internationale a été établie en 2017 ; elle identifie et décrit 13 types de SED. Ces différents types de SED n’exposent pas aux mêmes complications et leur pronostic et leur prise en charge sont donc différents : il est donc très important de les distinguer.
Un ensemble de critères cliniques majeurs et mineurs ont été définis pour chaque sous-type de SED.
Le diagnostic moléculaire permet de confirmer le diagnostic précis et donne des informations sur le profil de transmission héréditaire, le risque de récurrence et le pronostic, et il peut aider à la prise en charge. De plus, il permet la formation de cohortes homogènes à des fins de recherche et d'interventions thérapeutiques futures.
Seul le SED hypermobile (SEDh) n’a pas de base génétique connue, son diagnostic repose donc sur des critères cliniques, définis dans la classification de 2017. Si l’ensemble des critères nécessaires pour parler de SEDh ne sont pas remplis, le terme de pathologies du spectre de l’hypermobilité peut être utilisé, après avoir éliminé les diagnostics différentiels.
Le diagnostic se construit grâce à des éléments d’anamnèse personnels et familiaux. Il devrait être idéalement posé par des praticiens ayant acquis une expertise clinique au sein d’un centre de référence ou de compétences. Le diagnostic de certitude repose sur la confirmation génétique réalisée dans le cadre d’une consultation de génétique spécialisée dans les SED (sauf pour le SEDh, dont les bases génétiques ne sont pas encore identifiées).
Les principaux signes cliniques à rechercher sont : hyperlaxité articulaire (dépistée par le score de Beighton), entorses et luxations multiples, scoliose, troubles de la proprioception, douleurs articulaires, hyperextensibilité cutanée, difficultés de cicatrisation, hématomes anormaux, fragilité des tissus conjonctifs, fatigabilité anormale. Le bilan paraclinique initial recherchera un des nombreux diagnostics différentiels : maladie osseuse, neuromusculaire ou rhumatologique...
Le bilan biologique comprendra selon les patients les dosages suivants : bilan inflammatoire, bilan auto-immun, bilan d’hémostase, TSH, CPK, bilan phosphocalcique… Une radiographie de rachis complet et une échographie cardiaque doivent être réalisées de façon systématique (sauf contre-indication).
D’autres examens pourront être proposés par les spécialistes, en fonction des symptômes à la recherche de complications. Des bilans ophtalmologique, de kinésithérapie, de psychomotricité, d’ergothérapie et parfois orthophonique pourront être prescrits en fonction de la présentation clinique.
La prise en charge est complexe. Il n’existe pas de traitement spécifique mais une prise en charge pluridisciplinaire adaptée permet d’améliorer la qualité de vie des malades, en stabilisant l’évolution de la maladie et en prévenant ses complications.
L’annonce diagnostique est celle d’une maladie chronique et d’une maladie génétique, faisant souvent suite à une longue errance diagnostique. Une prise en charge psychologique personnalisée doit être systématiquement proposée dès l’annonce du diagnostic.
Un protocole national de diagnostic et de soins (PNDS) est disponible sur le site de la HAS. Son objectif est d’expliciter aux professionnels concernés la prise en charge diagnostique et thérapeutique optimale actuelle et le parcours de soins d’un patient atteint d’un des types de syndrome d’Ehlers-Danlos (SED) non vasculaire (SED NV). Il a pour but d’optimiser et d’harmoniser la prise en charge et le suivi de ces maladies rares sur l’ensemble du territoire. Il permet également d’identifier les spécialités pharmaceutiques, en particulier utilisées dans une indication non prévue dans leur autorisation de mise sur le marché (AMM), produits ou prestations nécessaires à la prise en charge des patients. Il est à noter que ces produits et prestations ne sont pas tous remboursés et que leur présence dans le PNDS ne conduit pas forcément à leur remboursement. Ce PNDS peut servir de référence au médecin traitant (médecin désigné par le patient auprès de la caisse d’assurance maladie) en concertation avec le médecin spécialiste notamment au moment d’établir le protocole de soins conjointement avec le médecin conseil et le patient, dans le cas d'une demande d'exonération du ticket modérateur au titre d'une affection hors liste.
Le PNDS ne peut cependant pas envisager tous les cas spécifiques, toutes les comorbidités ou complications, toutes les particularités thérapeutiques, tous les protocoles de soins hospitaliers, etc. Il ne peut pas revendiquer l’exhaustivité des conduites de prise en charge possibles, ni se substituer à la responsabilité individuelle du médecin vis-à-vis de son patient.
Des programmes d’Education Thérapeutiques Patients ont été validés par certaines Agences Régionales de Santé (Garches, Lyon, Toulouse).
Qu’est-ce qu’un syndrome d’Ehlers Danlos vasculaire ?
Le syndrome d’Ehlers Danlos vasculaire (anciennement type IV) est une maladie génétique rare (1 patient / 150 000) due à des mutations hétérozygotes du gène COL3A1 touchant le collagène de type III.
Sa transmission est autosomique dominante, non liée au sexe puisque le gène COL3A1 se situe sur le chromosome 2.
Comme la plupart des gènes, le gène COL3A1 est présent en 2 exemplaires dans notre génome. Il suffit qu’une des 2 copies soit défectueuse pour que la production de collagène de type III soit altérée et pour que la maladie soit possible.
Le collagène de type III est particulièrement présent dans les vaisseaux (artères et veines), les intestins, la peau, l'utérus, mais aussi dans les poumons, le foie, la rate et les capsules articulaires. Comment pose-t-on le diagnostic ? La maladie se diagnostique le plus souvent à l’occasion d’une complication vasculaire ou digestive. Elle peut aussi être retrouvée lors d’une recherche génétique familiale, lorsqu’un des membres de la famille est atteint.
Retrouver tous les autres types d’Ehlers-Danlos : unsed.org
Toutes les informations proviennent des critères de New York, du PNDS SEDNV et de la synthèse à destination des médecins traitants.
Pour se procurer des informations complémentaires, il est possible de consulter les sites :
- Union nationale des Syndromes d’Ehlers-Danlos (unsed) : unsed.org
- Le Centre de Référence des Syndromes d’Ehlers-Danlos non vasculaires (SED NV) de Garches : www.aphp.fr/service/service-51-068
- Filière de santé maladies rares FAVA-MULTI : www.favamulti.fr/pathologies-prises-en-charge/sedv/ #1442525650589-35e4494f-1b9c
- Centre de référence des maladies osseuses constitutionnelles (MOC) : syndromes d’Ehlers-Danlos non vasculaires (SED NV) : www.maladiesraresnecker. aphp.fr/sed/
- Centre de référence des maladies vasculaires rares (SEDv) : www.maladies-vasculaires-rares.fr/syndrome-dehlers-danlos-vasculaire
- Filière de santé maladies rares OSCAR : https://www.filiere-oscar.fr/ Orphanet : www.orphanet.net
Dossier rédigé par :
Docteur Karelle BENISTAN
Centre de référence des syndromes d’Ehlers-Danlos non vasculaires
Mme Valerie Gisclard
Présidente de l’UNSED (Union Nationale des Syndromes d’Ehlers-Danlos)
Expert Patiente, représentante des Usagers de Santé